The Nupam Mahajan Lab aims to determine how tyrosine kinases, phosphatases, demethylases and nuclear receptors regulate epigenetic processes that affect cellular homeostasis, immune impairment and cancer.
The lab is led by Nupam Mahajan, PhD, director of urologic research and Endowed Professor of Urologic Surgery Research at WashU Medicine.
Dr. Mahajan’s research focuses on the exploration of arious epigenetic processes to identify precise signaling events that take place in cancer cells, either during oncogenesis or upon exposure to various chemotherapeutic agents, including immune therapies.
The Nupam Mahajan Lab employs:
- ACK1 (TNK2): Immune modulator and oncogene
- WEE1: A novel epigenetic modulator
- SHP2 (PTPN11): Role in cancer and LEOPARD syndrome
- Androgen Receptor (AR): Prostate cancer and immunity
- KDM6A (UTX): Cancer & Multiple sclerosis
Principal investigator

Nupam Mahajan, PhD
Endowed Professor of Urologic Surgery Research
Director of Urologic Research
Division of Urologic Surgery
Section of Surgical Therapeutics
Division of Surgical Sciences
- Phone: 314-273-7759
Contact
PI Office:
Cancer Research Building, Room 6601
Campus Box 8242
660 S. Euclid Ave.
St. Louis, MO, 63110-1093
Lab Shipping Address:
Nupam Mahajan Lab
Cancer Research Building, Room 6606
Campus Box 8242
660 S. Euclid Ave.
St. Louis, MO, 63110-1093
E-mail: [email protected]
Phone:
Office: 314-273-7727
Lab: 314-273-7759
Fax: 314-272-7771
Opportunities
Graduate students
We are accepting graduate students from DBBS (Cancer Biology, Molecular Genetics & Genomics, Molecular Cell Biology, Human & statistical Genetics) and Dept of Biomedical Engineering.
Email: [email protected]
Postdoctoral opportunities
We are in recruitment drive. Send your CV to: [email protected]
Major findings
Discovered 7 new epigenetic events:
- Y88-Phosphorylation of histone H4
- Y37-Phosphorylation of Histone H2B
- Y54-Phosphorylation of Histone H3
- K130-acetylatylation of Histone H2A
- Y-Phosphorylation of histone H2A
- Y99-Phosphorylation of histone H3
Identified 3 new Inhibitors
- ACK1 inhibitor, (R)-9b: IND#167907
- WEE1 inhibitor
- KDM6A inhibitor
Identified a novel lncRNA, NXTAR
Identified AKT Tyr176-phosphorylation
Identified AR Lys609-acetylation
Current research
ACK1 activation in multiple subtypes of breast cancers
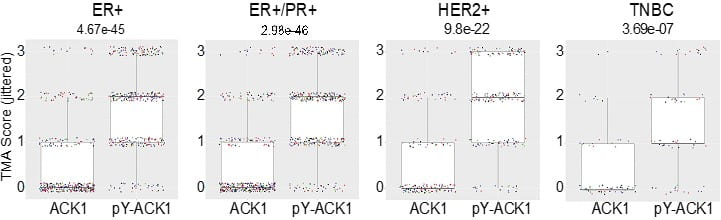
Immunohistochemical (IHC) analysis revealed a moderate to strong ACK1 activation in 58% of ER+/PR+, 70% of HER2+, and 47% of TNBC breast cancer samples (Figure 1).
Read more: Oncogene, 17 June, 2023.
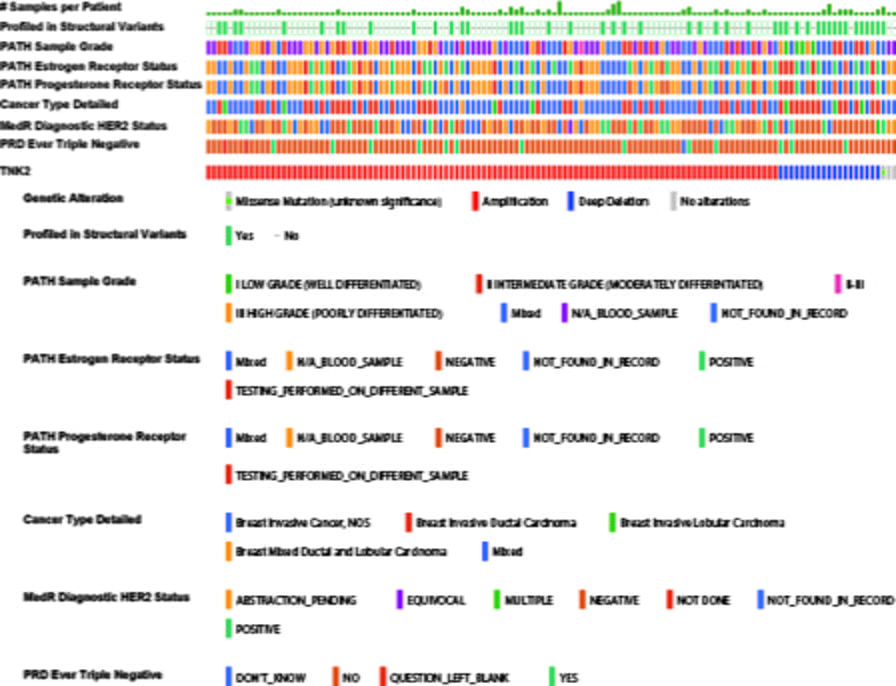
ACK1/TNK2 gene amplified in 30% of metastatic breast cancers
cBioPortal reveals that 30% of metastatic breast cancer exhibited ACK1/TNK2 gene amplification, indicating that the gene amplification as the major cause of the ACK1 activation in breast cancers (Figure 2). The ACK1 gene amplification was seen in:
- 20% of ER+
- 11% of ER+/PR+
- 13% of HER2+
- 9% of TNBCs (Figure 2).
Overall, a significant proportion of four distinct subtypes of breast cancers exhibit ACK1 activation, which is independent of their hormone receptor status.
ACK1 promotes Palbociclib resistance
ACK1 epigenetically regulates expression of cell cycle genes, CCNB1, CCNB2 and CDC20. Pharmacological inhibition of ACK1 using its inhibitor, (R)-9b dampened CCNB1, CCNB2 and CDC20 expression, causing G2/M arrest, culminating in regression of palbociclib-resistant breast tumor growth. Thus, (R)-9b could be a novel therapeutic option for the breast cancer patients those have developed resistance to CDK4/6 inhibitors.
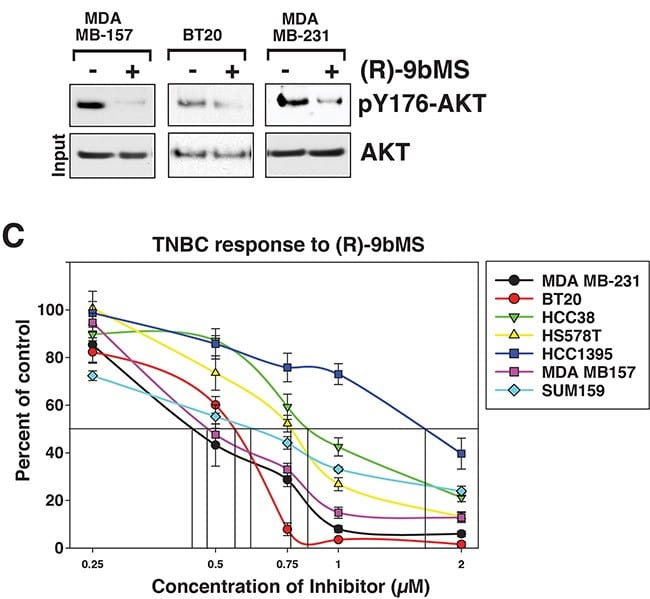
ACK1 as a new therapeutic target in TNBCs
About 15-20% of all breast cancers do not express estrogen, progesterone or HER2 receptor and classified as triple negative breast cancers (TNBC). These tumors are difficult to treat due to the lack of actionable target. We have uncovered that ACK1 is amplified and activated in TNBCs. Further, we show that genetic or pharmacological loss of ACK1 activity by its inhibitor (R)-9b can suppress breast tumor proliferation (Figure 3).
These data suggests that (R)-9b could be a new therapeutic strategy for breast cancers.
ACK1 activation in cancers
- (i) ACK1 integrates signals from various receptor tyrosine kinases, e.g. MERTK, AXL, EGFR, PDGFR, Insulin receptor, HER2. These kinases activate ACK1 in multiples cancers, including prostate, breast, lung, gastric, pancreatic, and many other cancers.
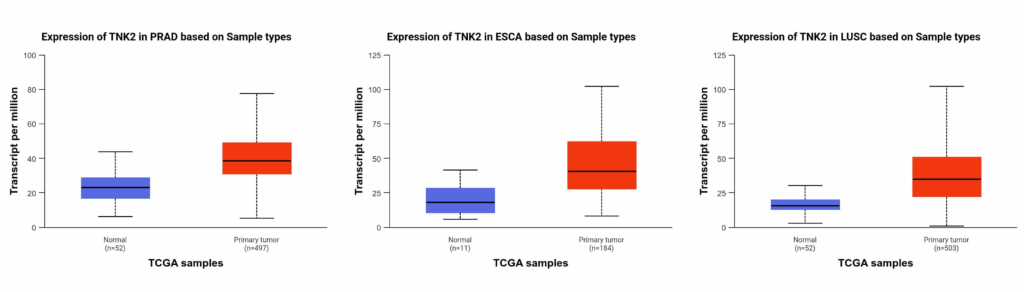
- (ii) ACK1/TNK2 gene is amplified in 30% of lung cancers, 20% of Urothelial carcinoma (bladder cancer), 20% of Esophageal carcinoma and 17% of ovarian cancers.
- (iii) A significant increase in ACK1 transcripts is also seen in prostate cancer, esophageal cancer and lung squamous cell carcinoma (Figure 3).
- (iv) Autoactivating mutations and gene fusions
Identification of ACK1 inhibitor, (R)-9b
No ACK1 inhibitor has so far made it to clinical trial. We generated a new class of small molecule ACK1 inhibitor (R)-9b that has excellent drug-like properties. (R)-9b suppressed proliferation of various prostate, breast and lung xenograft and PDX tumor growth.
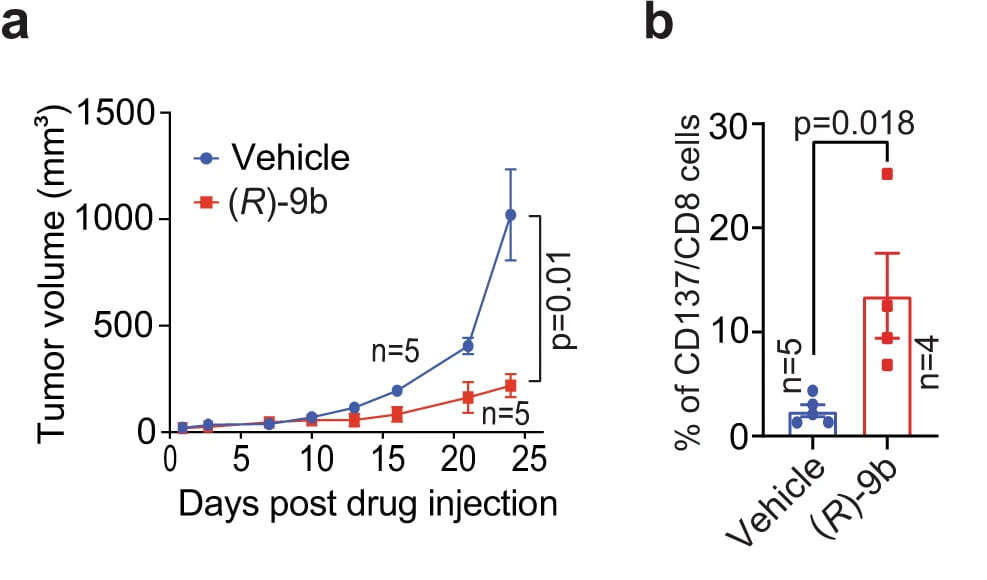
ACK1 inhibits syngeneic tumor growth by activating T cells
The B6 mice injected with (R)-9b exhibited a marked decrease in tumor growth (Figure 1). Harvested lymph nodes from the (R)-9b-treated mice revealed a significant increase in the number of CD137+/CD8+ cytotoxic T cells, effector CD44hiCD62Llow CD8+ T cells. Nature Communications, 2022.
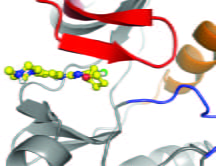
Crystal structure of (R)-9b bound to ACK1
We have shown the crystal structure of (R)-9b bound to ACK1 kinase domain. Nature Communications, 2022.
Clinical Trial of ACK1 inhibitor, (R)-9b
Phase I Clinical trial (PHAROS) of (R)-9b is expected to start in early 2026 (IND#167907). Dr. Nupam Mahajan ([email protected]) can be contacted by interested parties.
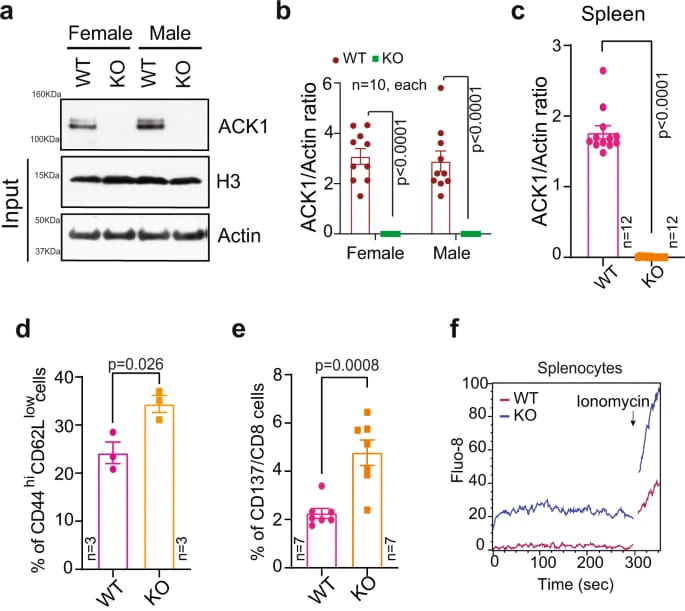
ACK1 knockout mouse
T-cell activation is negatively regulated by C-terminal Src kinase (CSK). We uncovered that ACK1 phosphorylated CSK at Tyrosine 18 (pY18), which enhanced CSK function, constraining T-cell activation. Mice deficient in the Ack1/Tnk2 gene are characterized by diminished CSK Y18-phosphorylation and spontaneous activation of CD8+ and CD4+ T cells (Figure 1), resulting in inhibited growth of transplanted ICB-resistant tumors. Furthermore, ICB treatment of castration-resistant prostate cancer (CRPC) patients results in re-activation of ACK1/pY18-CSK signaling, confirming the involvement of this pathway in ICB insensitivity. Nature Communications, 2022.
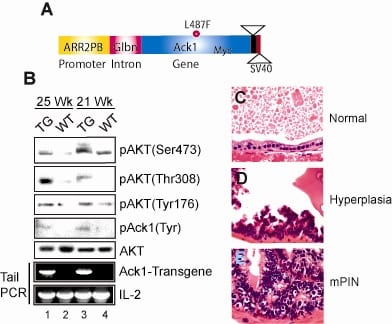
ACK1 transgenic mouse
To understand the pathophysiological role of ACK1 signaling, we generated a transgenic mouse model in which Myc-tagged activated ACK1 was expressed under the control of modified Probasin (PB) promoter, ARR2PB (Figure 2A). These PB-ACK1 transgenic mice display significant increase in AKT Tyr176-phosphorylation leading to Ser473/Thr308-phosphorylation in prostates (Figure 2B, top 3 panels). These mice developed prostatic intraepithelial neoplasia or mPINs (Figure 2E). Pubmed ID: 20333297
Thus, ACK1 activation could be an independent tumor initiation event in those cancer patients that exhibit normal PTEN and PI3K, but still exhibit AKT activation. Pubmed ID: 20333297
The AKT/PKB kinase is activated by PI3K activation or loss of PTEN. About a third of breast and prostate tumors and majority of the pancreatic tumors that exhibit AKT activation retain normal PTEN and PI3K activity.
Novel AKT Tyr176-phosphorylation, mediated by ACK1
(Pubmed ID: 20333297)
ACK1 directly phosphorylated AKT at Tyr176 (Figure 1). For membrane localization, AKT anchors to Phosphatidylinositol (3,4,5)-trisphosphate or PIP3. However, Tyr176-phosphorylated AKT bound another membrane phospholipid, phosphatidic acid (PA). Further, Tyr176-phosphorylated AKT translocated to the membrane leading to its Ser473-phosphorylation and kinase activation. Pubmed ID: 20333297
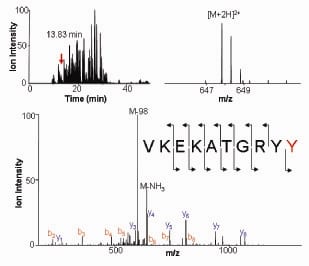
AKT Tyr176-phosphorylation in Prostate and breast cancers
We generated ACK1 transgenic mice that not only exhibited AKT Tyr176-phosphorylation but also developed prostatic intraepithelial neoplasia (PINs) and rare carcinomas. Pubmed ID: 20333297
The most significant role of pY176-AKT was observed in progression of breast cancer (Figure 2A). The expression levels of pY176-AKT and activated ACK1 were positively correlated with the severity of breast cancer progression (Figure 2B and C), and inversely correlated with the survival of patients (Figure 2D and E). Pubmed ID: 20333297
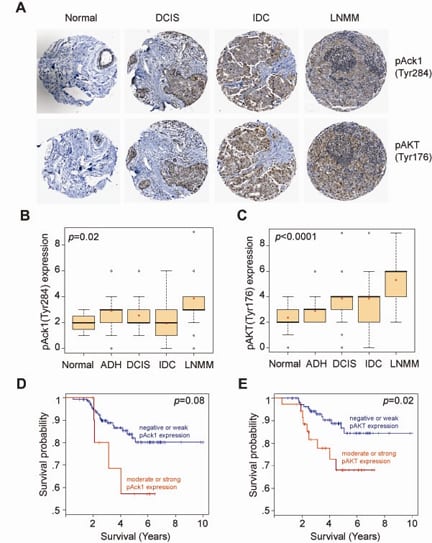
Solid tumors are highly refractory to immune checkpoint blockade (ICB) therapies due to the functional impairment of effector T cells and their inefficient trafficking to tumors. T-cell activation is negatively regulated by C-terminal Src kinase (CSK); however, the exact mechanism remains unknown. We uncovered that ACK1 phosphorylate CSK at Tyrosine 18 (pY18), which enhances CSK function, constraining T-cell activation. Mice deficient in the ACK1/Tnk2 exhibit spontaneous activation of CD8+ and CD4+ T cells, resulting in inhibited growth of transplanted ICB-resistant tumors.
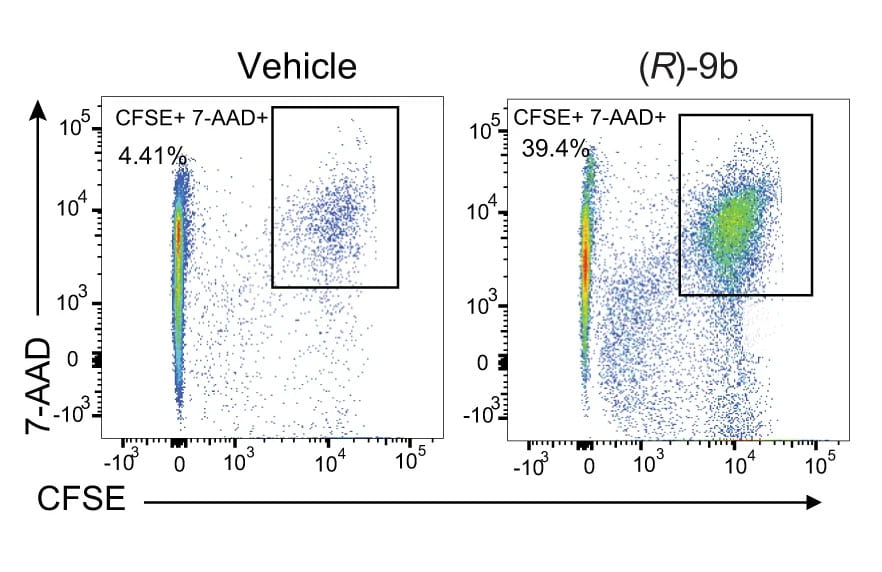
Splenocytes from C57BL/6 mice were treated with (R)-9b. After the compound was washed off, a cell-killing assay was performed wherein CFSE-stained TRAMP-C2 cells were incubated with splenocytes. A significant increase in TRAMP-C2 cell death (CFSE+ 7-AAD+ cells) was observed for the splenocytes treated with (R)-9b compared with those treated with vehicle (Figure 1). Nature Communications, 2022.
ACK1-AR signaling in prostate cancer
ACK1 (TNK2) interacts with androgen receptor or AR in an androgen-independent manner. Expression of activated ACK1 correlates positively with the progression of disease to CRPC stage, and PC patients whose tumors display moderate to strong staining of activated ACK1 have poor prognosis.
AR plays a paramount role in the onset and progression of prostate cancer (PC). A majority of the PC patients progress to a lethal stage of the disease, referred to as the metastatic Castration Resistant Prostate Cancer (mCRPC). There is increasing expression of AR as disease progress to later stages (Figure 2). CRPC remains an incurable malignancy with limited treatment options.
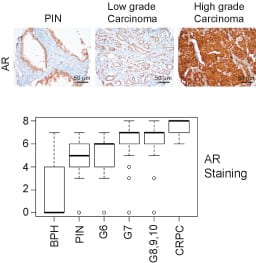
Need to venture beyond Anti-Androgen Therapies
Reliance of CRPCs on AR despite androgen-depletion, has led to development of AR antagonists, Enzalutamide & Abiraterone. Unfortunately, in spite of early response, most patients relapsed within 2 years, exhibiting renewed AR activity.
ACK1, a new target in CRPCs
ACK1 is upregulated in ~40% of prostate adenocarcinomas. Importantly, 10 out of 13 CRPCs exhibited ACK1 overexpression. Further, LNCaP cells that are poorly tumorigenic in castrated mice, formed robust CRPC tumors following expression of activated ACK1.
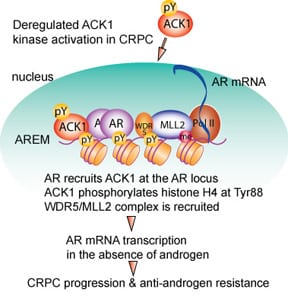
ACK1, a histone kinase
We uncovered that ACK1 interacts with AR and deposit novel epigenetic marks, histone H4 Tyr88-phosphorylation (Figure 3). Inhibition of ACK1 by (R)-9b suppressed AR & AR-V7 transcription, inhibiting proliferation of enzalutamide-resistant CRPC tumors.
Mahajan K, Malla P, Kim J, Coppola D, Lawrence N and Mahajan NP*. ACK1 regulates histone Tyr-phosphorylation and AR gene expression in castration resistant prostate cancer. Cancer Cell 2017. Pubmed ID: 28609657
KDM6A inhibitor
Lysine-specific demethylase 6A (KDM6A) gene encodes a protein known as Ubiquitously transcribed tetratricopeptide repeat protein on X chromosome (UTX). The KDM6A gene is conserved and located on the X-chromosome in both mice, and humans. KDM6A belongs to the 2-oxoglutarate (2OG)-dependent dioxygenase superfamily and catalyzes demethylation of H3K27me3 epigenetic marks causing transcriptional activation. Circulating tumor cell (CTC) exhibited gain of KDM6A upon Abi and Enz treatment, and mutations in KDM6A were identified in high grade prostatic intraepithelial neoplasia (HGPIN) that preceded prostate cancer. Heavily-pretreated metastatic CRPCs exhibited KDM6A gene copy number gain in these patients. Further, SU2C dataset revealed that 9.3% of prostate cancers exhibits KDM6A gene amplification, suggesting that its enhanced expression promotes prostate cancer.
We designed an assay and the library screen identified a new class of scaffolds, that efficiently suppressed KDM6A epigenetic activity and have shown to overcome prostate, breast, and cervical cancer cell proliferation and tumor growth.
KDM6A is overexpressed in prostate cancers, triple negative breast cancer, pancreatic cancer, esophageal cancer, Head and Neck Cancer, Glioblastoma and Mesothelioma.
Feel free to contact us if you are interested in exploring these novel inhibitors in your lab.
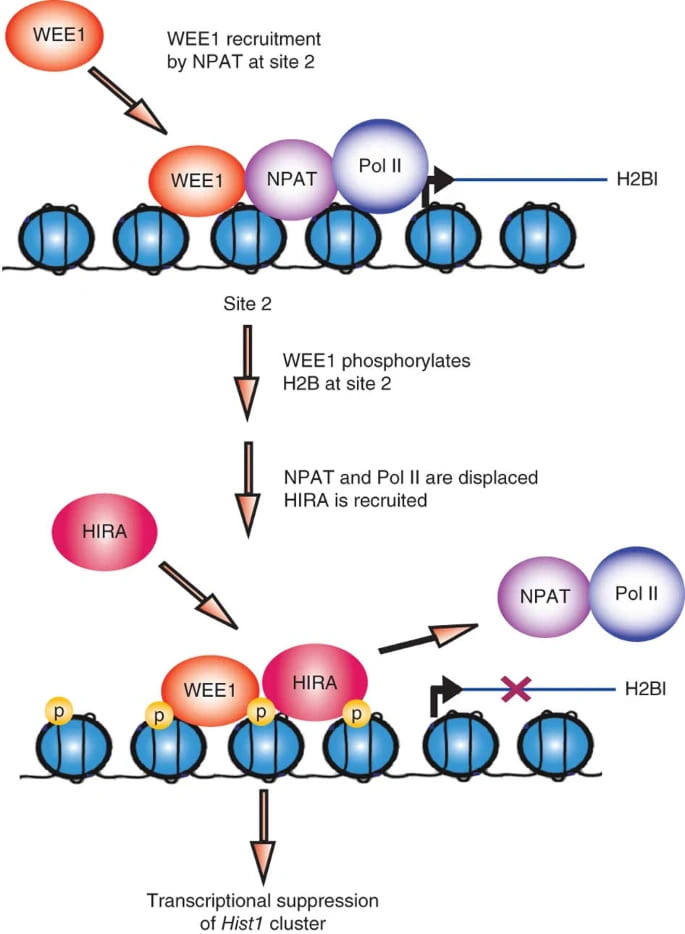
A Novel Epigenetic Function of WEE1
We discovered that WEE1 kinase phosphorylates histone H2B at Tyr37 (py37-H2B) in a window of 30-40 minutes in late S phase. Eukaryotic cells precisely regulates histone levels by shutting off histone transcription at the end of DNA synthesis. WEE1 kinase deposit pY37-H2B epigenetic marks upstream of Hist1 cluster, suppressing global histone transcription in both yeast and mammalian cells.
Histone transcription shut off would efficiently lower histone transcript levels once DNA synthesis is complete, eliminating overproduction of core histones.
Mahajan K, Fang B, Koomen JM and Mahajan NP*. H2B Tyr37 phosphorylation suppresses expression of replication-dependent core histone genes. Nature Structural & Molecular Biology, 2012
WEE1 epigenetically regulates 5hmC levels
WEE1 kinase has been reported to be aberrantly expressed in melanomas, glioblastoma multiforme (GBMs), triple negative breast cancers (TNBCs) and prostate cancer. Dependence of various cancer cells on WEE1 signaling suggests that targeting epigenetic activity of WEE1 is a viable strategy.
WEE1 Inhibitor in Prostate cancer, Melanoma and GBM
WEE1-H2B epigenetic signaling plays a crucial role in various malignancies such as melanoma, GBM and prostate cancer
Our team
Nupam Mahajan, PhD
Endowed Professor of Urologic Surgery Research
Director of Urologic Research
Division of Urologic Surgery
Section of Surgical Therapeutics
Division of Surgical Sciences
- Phone: 314-273-7759
Staff
Elliot Bradshaw, PhD
Staff Scientist
Dhivya Sridaran, PhD
Staff Scientist
Jagat Krishna Shrestha, PhD
Postdoctoral Research Associate
Eswara Rao Puppala, PhD
Postdoctoral Research Associate
Shalini Gupta, PhD
Postdoctoral Research Associate
Samiksha Yogi
Research Associate
Mao Yoshikawa-Kawana, MD, PhD
Postdoctoral Research Associate


